
Jakarta – Para peneliti memetakan beberapa penyebaran asli virus corona pada manusia dan menemukan ada beberapa varian virus di seluruh dunia. Mereka merekonstruksi jalur evolusi awal Covid-19 ketika infeksi menyebar dari Wuhan, Cina, ke Eropa dan Amerika Utara.
Dengan menganalisis 160 gen virus lengkap pertama yang diurutkan dari pasien manusia, para ilmuwan menemukan varian yang paling dekat dengan yang ditemukan pada kelelawar, sebagian besar ditemukan pada pasien dari AS dan Australia, bukan Wuhan.
Dr Peter Forster, ahli genetika dan penulis utama dari University of Cambridge, seperti dikutip dari Metro.co.uk, mengatakan, terlalu banyak mutasi yang cepat untuk melacak silsilah keluarga Covid-19 dengan rapi.
“Kami menggunakan algoritma jaringan matematika untuk memvisualisasikan semua keluarga virus ini yang masuk akal secara bersamaan. Teknik-teknik ini sebagian besar dikenal untuk memetakan pergerakan populasi manusia prasejarah melalui DNA. Kami pikir ini adalah salah satu pertama kalinya mereka digunakan untuk melacak rute infeksi virus corona seperti Covid-19,” katanya, Jumat (10/4/2020).
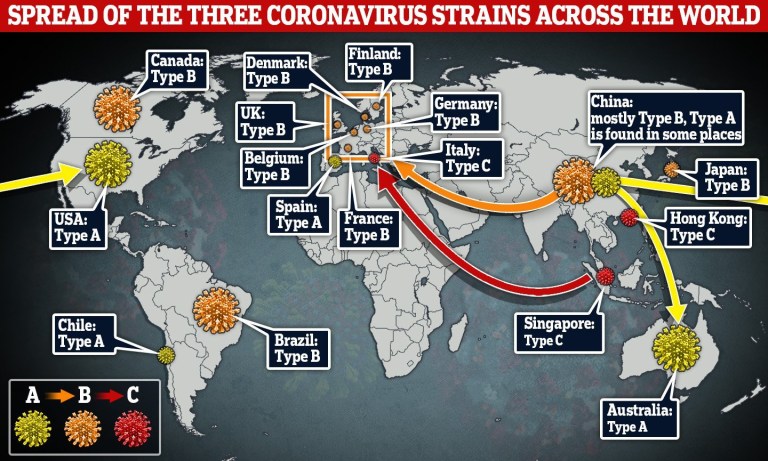
Tim menggunakan data dari sampel yang diambil dari seluruh dunia antara 24 Desember 2019 dan 4 Maret 2020. Mereka menemukan tiga varian Covid-19 yang berbeda, tetapi berkaitan erat, yang mereka sebut A, B dan C.
Para peneliti menemukan bahwa jenis virus corona yang paling dekat dengan yang ditemukan pada kelelawar – tipe A, genom virus manusia asli – ada di Wuhan, tetapi bukan jenis virus utama di kota itu.
Versi mutasi A terlihat di Amerika yang dilaporkan telah tinggal di Wuhan, dan sejumlah besar virus tipe A ditemukan pada pasien dari AS dan Australia.
Jenis virus utama Wuhan adalah B dan lazim pada pasien dari seluruh Asia timur, namun tidak banyak bepergian di luar kawasan tanpa mutasi lebih lanjut.
Para peneliti mengatakan varian C adalah tipe Eropa utama, ditemukan pada pasien awal dari Perancis, Italia, Swedia dan Inggris.
Analisis ini juga menunjukkan salah satu perkenalan awal virus ke Italia datang melalui infeksi Jerman yang pertama kali didokumentasikan pada 27 Januari, dan bahwa rute infeksi Italia awal lainnya terkait dengan ‘cluster Singapura’.
Itu tidak ada dalam sampel daratan Cina studi ini tetapi terlihat di Singapura, Hong Kong, dan Korea Selatan. Para ilmuwan berpendapat metode mereka dapat diterapkan pada sekuensing genom coronavirus terbaru untuk membantu memprediksi hotspot global masa depan dari penularan dan penyebaran penyakit.
Temuan ini diterbitkan dalam jurnal Proceeding of National Academy of Sciences (PNAS). Varian A, yang paling dekat hubungannya dengan virus yang ditemukan pada kelelawar dan trenggiling, digambarkan sebagai akar wabah oleh para peneliti.
Tipe B berasal dari A, dipisahkan oleh dua mutasi, kemudian C pada gilirannya adalah ‘anak perempuan’ dari B, penelitian menunjukkan. Metode jaringan filogenetik yang digunakan oleh para peneliti – yang melihat hubungan evolusi di antara entitas biologis – memungkinkan visualisasi ratusan pohon evolusi secara bersamaan dalam satu grafik sederhana. [Zin]






